Comparing Runtime across Sample Sizes and Number of Taxa
Source:vignettes/articles/runtime_evaluation.Rmd
runtime_evaluation.RmdIntroduction
This vignette studies how phylobar’s runtime scales with the number
of samples (N) and taxa (P). We simulate random data across a grid of
(N, P) values and time each call with system.time(). We
also compare runtime with and without ordering samples with hierarchical
clustering (the hclust_order argument).
Setup
We simulate one large dataset at the maximum grid dimensions and subset it for each (N, P) combination. This avoids repeated simulation and naming boilerplate inside the timing loops.
n_eval <- c(50, 100, 250, 500, 1000, 2000)
p_eval <- c(50, 100, 250, 500, 1000)
n_reps <- 10
grid <- expand.grid(
N = n_eval,
P = p_eval,
hclust_order = c(TRUE, FALSE),
rep = seq_len(n_reps)
)
tree_full <- rtree(max(p_eval))
x_full <- matrix(
rpois(max(n_eval) * max(p_eval), lambda = 5),
nrow = max(n_eval), ncol = max(p_eval)
)
colnames(x_full) <- tree_full$tip.label
rownames(x_full) <- paste0("s", seq_len(max(n_eval)))
trees <- list()
for (p in p_eval) {
tips <- tree_full$tip.label[seq_len(p)]
trees[[as.character(p)]] <- drop.tip(
tree_full, setdiff(tree_full$tip.label, tips)
)
}Timing
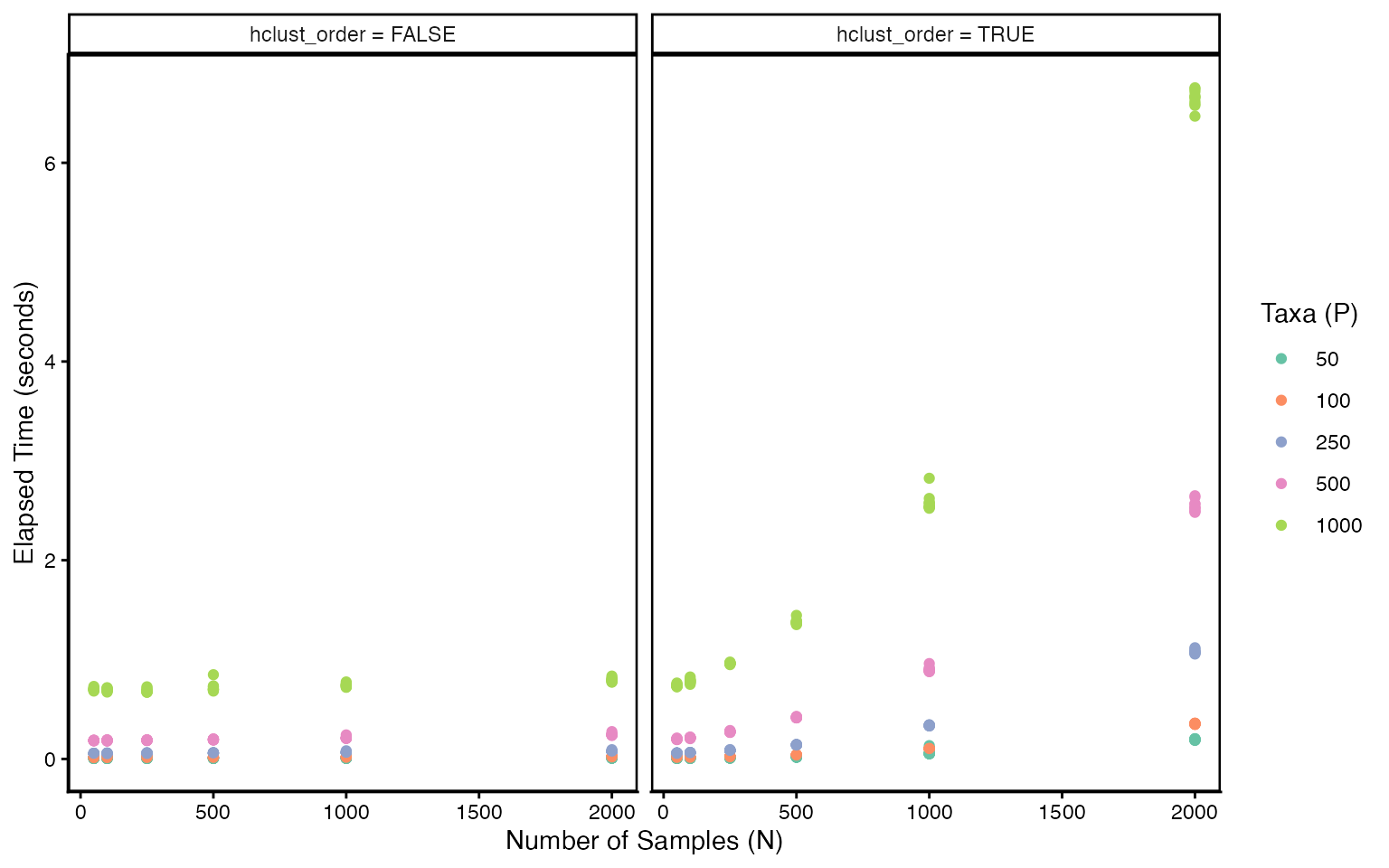
For each (N, P, hclust_order) combination we subset the pre-simulated
data and time phylobar(). By default phylobar
sorts samples using hierarchical clustering, which can be costly when
the number of samples is large. It is also often unnecessary when there
is already a natural sample ordering (e.g., over time). Setting
hclust_order = FALSE keeps samples in their input
order.
results <- grid
results$elapsed <- NA
for (i in seq_len(nrow(results))) {
tree <- trees[[as.character(results$P[i])]]
x <- x_full[seq_len(results$N[i]), tree$tip.label]
tm <- system.time(
phylobar(x, tree, hclust_order = results$hclust_order[i])
)
results$elapsed[i] <- tm["elapsed"]
}Results
results$P <- factor(results$P)
results$hclust_label <- ifelse(
results$hclust_order,
"hclust_order = TRUE",
"hclust_order = FALSE"
)
ggplot(results, aes(N, elapsed, color = P)) +
geom_point() +
scale_color_brewer(palette = "Set2") +
facet_wrap(~ hclust_label) +
theme(panel.border = element_rect(fill = NA, linewidth = 0.9)) +
labs(
x = "Number of Samples (N)",
y = "Elapsed Time (seconds)",
color = "Taxa (P)"
)
Visualization
Here is an example of the phylobar visualization made with
samples and
taxa. While phylobar generates the plot SVG elements in a
few seconds, the output is both difficult both for the browser to render
interactively and for us to visually interperet. We’ve disabled this
plot in this documentation because it consumes quite a lot of browser
memory drawing all the bars in the barplot.
# very slow rendering, not recommended
phylobar(x, tree, hclust_order = results$hclust_order[i])In this regime, we recommend using the subset_cluster
function to select representative samples to include in the stacked bar
plot. Alternatively, separate phylobar views can be made for different
sample subsets. As long as we reduce
,
we can keep
and still have smooth interaction.
x_sub <- subset_cluster(x, k = 100)
x_sub <- x_sub[, colSums(x_sub) > 0]
phylobar(x_sub, tree, hclust_order = results$hclust_order[i])Takeaways
Bypassing the hierarchical clustering step helps in the large and setting. When the data are large, it may be worth using a custom, more scalable seriation algorithm to sort the rows of
xin advance, and then sethclust_order = FALSE.While the
phylobarfunction executes quickly, rendering and visual interpretation both suffer when is large. When and are large, the initial view will show rectangles in the stacked barplot. Rendering all rectangles as SVG elements in the browser can be slow, and stacked bar plots with too many bars are illegible. We recommend either finding representative samples or breaking the exploration into separate subsets of samples.
Session Info
sessionInfo()
#> R version 4.6.1 (2026-06-24)
#> Platform: x86_64-pc-linux-gnu
#> Running under: Ubuntu 24.04.4 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
#> [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
#> [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
#> [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] phylobar_0.99.12 ggplot2_4.0.3 ape_5.8-1 BiocStyle_2.40.0
#>
#> loaded via a namespace (and not attached):
#> [1] sass_0.4.10 generics_0.1.4 lattice_0.22-9
#> [4] digest_0.6.39 magrittr_2.0.5 evaluate_1.0.5
#> [7] grid_4.6.1 RColorBrewer_1.1-3 bookdown_0.47
#> [10] fastmap_1.2.0 jsonlite_2.0.0 Matrix_1.7-5
#> [13] BiocManager_1.30.27 purrr_1.2.2 scales_1.4.0
#> [16] codetools_0.2-20 textshaping_1.0.5 jquerylib_0.1.4
#> [19] cli_3.6.6 rlang_1.2.0 withr_3.0.3
#> [22] cachem_1.1.0 yaml_2.3.12 otel_0.2.0
#> [25] tools_4.6.1 parallel_4.6.1 dplyr_1.2.1
#> [28] fastmatch_1.1-8 vctrs_0.7.3 R6_2.6.1
#> [31] lifecycle_1.0.5 fs_2.1.0 htmlwidgets_1.6.4
#> [34] ragg_1.5.2 cluster_2.1.8.2 pkgconfig_2.0.3
#> [37] desc_1.4.3 pkgdown_2.2.0 bslib_0.11.0
#> [40] pillar_1.11.1 gtable_0.3.6 glue_1.8.1
#> [43] phangorn_2.12.1 Rcpp_1.1.1-1.1 systemfonts_1.3.2
#> [46] xfun_0.59 tibble_3.3.1 tidyselect_1.2.1
#> [49] knitr_1.51 farver_2.1.2 htmltools_0.5.9
#> [52] nlme_3.1-169 igraph_2.3.3 labeling_0.4.3
#> [55] rmarkdown_2.31 compiler_4.6.1 quadprog_1.5-8
#> [58] S7_0.2.2